Protein Homeostasis and Dynamics Group
We are a molecular systems biology lab focused on understanding protein dynamics and cellular processes through proteomics and bioinformatics approaches.
Research Focus
We take an interdisciplinary approach that combines proteomics technology with biological inquiries
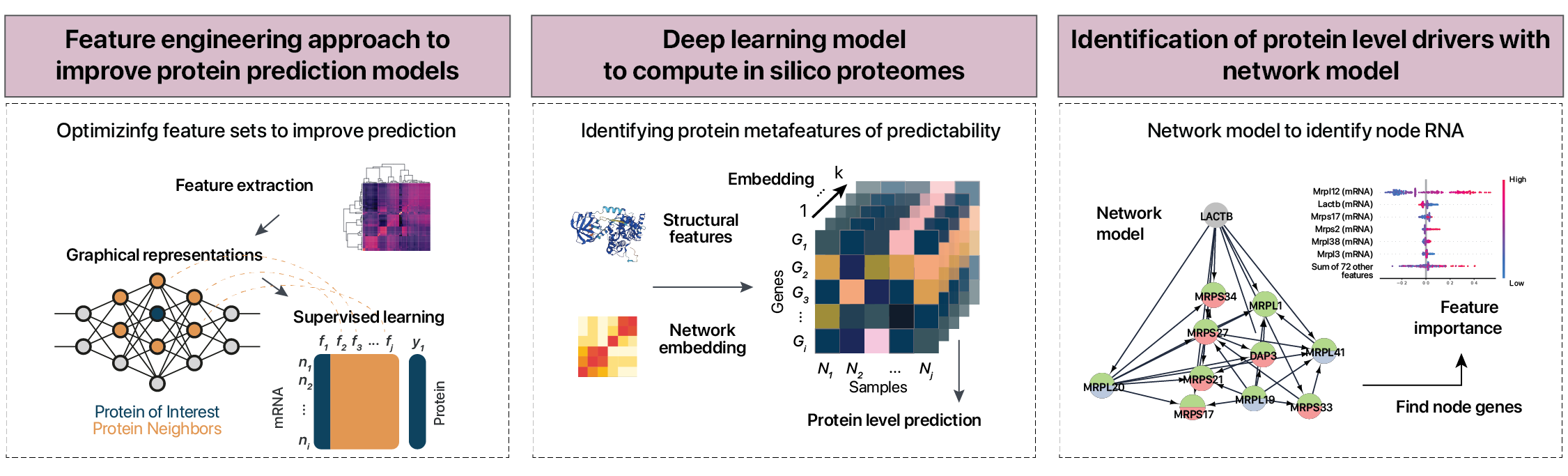
Predicting RNA-protein relationships
Machine learning and deep learning methods to predict proteins from mRNA
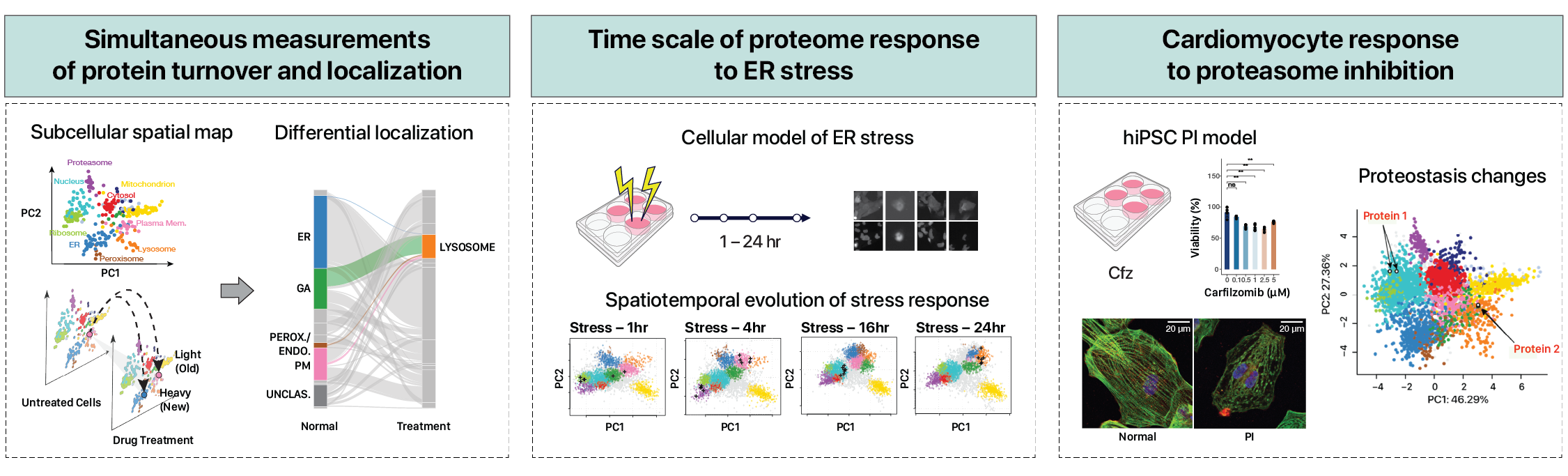
Spatiotemporal proteomics
Understanding the subcellular regulations of protein homeostasis
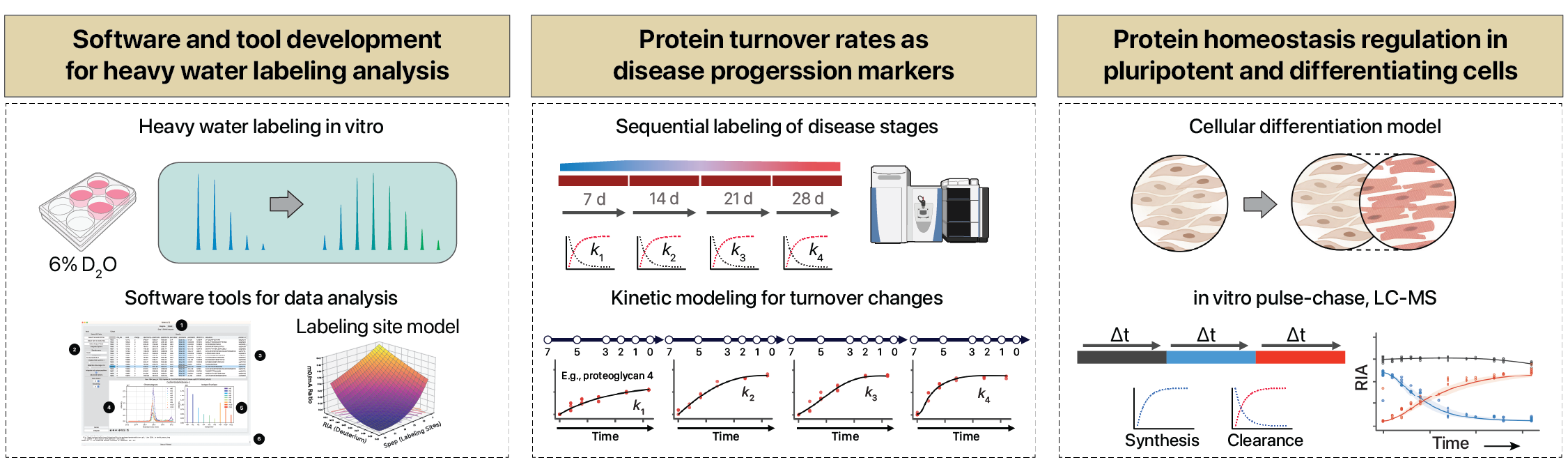
Protein turnover kinetics
We develop methods to measure protein synthesis and degradation flux
Featured Publications
Recent highlights from our research
SALVE: prediction of interorgan communication with transcriptome latent space representation.
Am J Physiol Heart Circ Physiol (2025)
Massive transcriptomics data allow gene relationships to be discovered from their correlated expression. We describe secretome association with latent variables …
Deuterium labeling enables proteome-wide turnover kinetics analysis in cell culture.
Cell Rep Methods (2025)
Protein turnover is a critical component of gene expression regulation and cellular homeostasis, yet methods for measuring turnover rates that …
Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of …
Nat Commun (2024)
The spatial and temporal distributions of proteins are critical to protein function, but cannot be directly assessed by measuring protein …

Principal Investigator
Edward Lau
Ed Lau is an Associate Professor in the Department of Medicine of the University of Colorado School of Medicine. He is also a faculty member of the Integrated Physiology, Pharmacology & Molecular Medicine, and Cell, Stem Cell & Development PhD programs on campus. Ed received his BA degree in molecular …
Research Interests: Protein turnover, kinetic models
Data & Tools
Open resources from our laboratory
Interactive Data
Lab Updates
Recent news and announcements
Follow Us on Bluesky
The process of experimental science does not consist in explaining the unknown by the known, as in certain mathematical proofs, it aims to give an account of what is observed by the properties of what is imagined.